
We Are Here To Help

We’re glad you found us. A diagnosis of KCNQ2-related epilepsy can be a confusing, even overwhelming step in your journey as a parent. There seem to be more questions than answers, all of which revolve around a central one: What does this mean for my child?
The good news is that dedicated researchers, doctors, and families are blazing a trail through this jungle of questions. Together, we are making progress. Below you’ll find some of the FAQs of KCNQ2. We hope they serve to guide you as you continue your journey forward.
Go at your own pace. We are here to help: info@kcnq2.org.
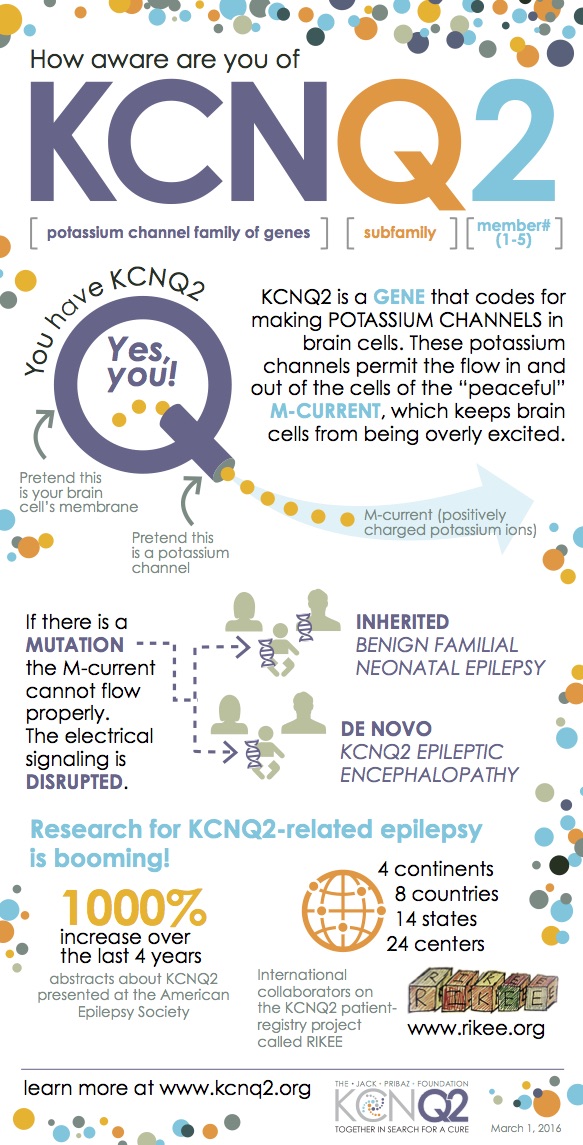
KCNQ2 is a gene involved in the proper functioning of a potassium channel in the brain. Abnormal changes, or mutations, in the gene are associated with seizures. KCNQ2-related epilepsies represent a spectrum of conditions from mild to severe. In 1998, mutations in KCNQ2 were associated with a mild condition called “Benign Familial Neonatal Epilepsy” or BFNE. Babies with BFNE have seizures that begin shortly after birth and then stop within several months. Development is usually normal. BFNE may run in families, as the name implies.
Researchers recently identified different mutations in KCNQ2 that are associated with a severe form of neonatal epilepsy. This severe, non-inherited (de novo) form has been called several names: KCNQ2 encephalopathy, KCNQ2-related neonatal epileptic encephalopathy, KCNQ2-deficiency syndrome, and KCNQ2-related epilepsy. As more patients are diagnosed with this emerging condition, doctors are finding that this mutation contributes to a spectrum of clinical outcomes that includes developmental problems ranging from moderate to severe.
Handy KCNQ2 Infographic
A Computer Model of KCNQ2

Find your local KCNQ2 Group – Search Now
Sign Up with the Facebook Community
View Google Scholar research publications