

Li Li, MD, PhD
Bringing KCNQ2 to a big stage
Do you ever wonder what it really means to “support research” for KCNQ2? In our first blog, we are happy to introduce you to a special person whose work was one of our earliest investments in KCNQ2 research.
Her abstract, “KCNQ2 encephalopathy: novel single amino acid deletion variants strongly suppress currents and are responsive to SF0034” was selected for platform presentation at the Pediatric Epilepsy Highlights Session at the American Epilepsy Society (AES) meeting in Philadelphia.
JPF: Please tell us a little bit about what motivates you in your research.
LL: Mutations in the potassium channel gene KCNQ2 cause medically refractory neonatal-onset epilepsy, global developmental delay, and autism. Increasing numbers of clinical cases of KCNQ2 encephalopathy in children have been found in the past couple of years. These cases encourage me to join KCNQ2 study groups.
As a mother to two boys, nothing makes me happier than seeing them smile, watching them laugh, play and grow up healthy. So I understand how hard it is when parents have to face something as hard as watching their child struggle in their development. Being a mother encourages me and gives importance to my goal: To discover good treatments for kids’ diseases using my knowledge and techniques.
.
JPF: Can you please summarize your abstract in lay terms?
LL: Our lab and collaborators have established an international registry of KCNQ2 cases from patients. To date, we have collected around 120 KCNQ2 encephalopathy missense variants and two interesting examples of single amino acid deletion. One is a unique variant (found in one case), the other in 6 children. My goal was to clarify whether these deletion variants were likely to be the cause of the seizures and impaired development seen in patients.
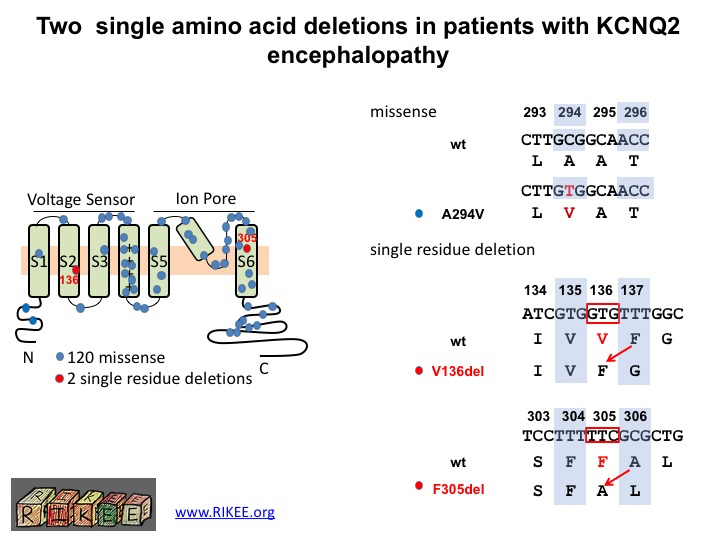
This diagram shows the location in the KCNQ2 protein of the two newly discovered single amino acid deletions (dots in red) and the 120 previously characterized missense mutations (dots in blue). A missense mutation is a point mutation in which a single nucleotide change results in a codon (a triplet of nucleotides) that codes for a different amino acid. A single amino acid deletion occurs when an entire codon is lost and the rest of the sequence is shifted. Dr. Li’s paper was the first to show that a single amino acid lost, not changed, can induce encephalopathic epilepsy.
JPF: Why do you think your abstract was selected as a highlight? What about your discovery is most valuable in advancing KCNQ2 (or epilepsy) research?
LL: I think my abstract was selected because it is the first time deletion variants have been shown to induce KCNQ2 encephalopathy. I detected strongly reduced channel function and showed a drug could reverse some of the effects of the variant on the channel.
JPF: When did you find out your abstract was selected for special presentation, and how did you prepare for the talk?
LL: In June, I submitted my abstract to the AES annual meeting. After 3 months, in September, I got an email from 2015 the AES scientific program committee. My abstract was selected for a Pediatric Epilepsy Highlights presentation. To prepare, Dr. Cooper and my coworkers gave a lot of valuable advice. It took around a month to organize our clinical and electrophysiological data, and I practiced many times before the presentation.
JPF: Describe your experience of presenting the work. What kind of feedback or questions did you get?
LL: About a thousand people attended my presentation. Most of the audience were doctors. They were very interested in the mechanism and drug treatment. Most of the questions I received were about drug side effects and rules to use drugs to treat encephalopathy, since I presented a new drug (SF0034) that showed similar effects as ezogabine (an FDA approved drug to treat seizures) but which may be less likely to cause the side effects of blue skin discoloration.
JPF: What are you working on next?
LL: I will try to complete these deletion variant studies and publish a paper in the near future. Also I will study other variants in KCNQ2 encephalopathy.

Left to right: Baouyen Tran, a PhD student, Li Li, and Jacquenae Mays, a post-Bac student. Jacquenae and Bao are Li’s collaborators and she thanks them for tirelessly helping her in preparation and rehearsal of her talk.
A native of Dalian, China, Dr. Li Li received her PhD in Department of Pharmacology and Neurobiology at Tokyo Medical and Dental University and her Medical Bachelor degree at Dalian Medical University in Dalian, Liaoning, China. She has studied ion channels for 10 years and joined Dr. Ed Cooper’s Lab at Baylor College of Medicine in 2012 as a postdoctoral research fellow. Her focus is to clarify the mechanisms underlying potassium and calcium channel diseases and develop new drug treatments to cure them
DONATE to The Jack Pribaz Foundation to support research.