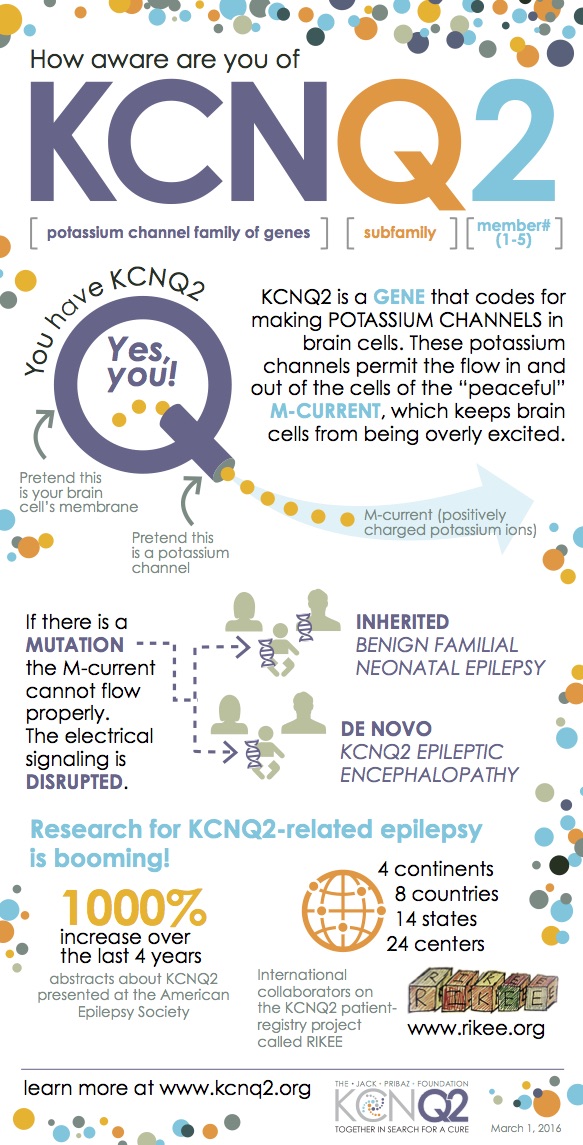
You probably know by now that in the gene name KCNQ2, K is for potassium and CN is for channel. But what about Q2 (and 1, 3, 4, and 5)? You might want to go to Italy to find out! The KCNQ family (aka Kv7) will have a sort of “reunion” this week at the International Kv7 Channel Symposium in Naples. Researchers from the across Europe, the United States (hello, Dr. Ed Cooper!), Australia, and China will gather to focus on their common ties and the ways they can help each other out for the benefit of many.
Download powerpoint slides for the meeting here!
Dr. Maurizio Taglialatela, MD, PhD, professor of pharmacology at the University of Naples Federico II and member of the medical advisory board for the Jack Pribaz Foundation, will host his colleagues in his hometown. “We hope to merge together in this meeting the best work done in the areas of neuroscience, cardiac, and muscle,” Taglialatela says, “so that cross-fertilization among disciplines might help in promoting better research and therapeutic opportunities for our patients.”
Recently Gina Pribaz Vozenilek interviewed Dr. Maurizio Taglialatela by email about the goals of the conference and the importance of considering the Kv7 genes together.
GPV: Can you explain the family of Kv7 genes and how they are related to one another?
MT: Among ~ 80 potassium channel genes found in humans, those belonging to the Kv7 family regulate the activity of cells in many organs including brain, heart and muscle. The five known Kv7 family members (Kv7.1-5 or KCNQ1-5) show distinct cellular and subcellular localization in each cell and often cooperate functionally with each other. For instance, KCNQ1 is mainly found in the heart whereas KCNQ2 and KCNQ3 are more abundant in the brain. Alteration of their function can cause diseases such as epilepsy, cardiac arrhythmias, or deafness, depending on their tissue- and time-specific expression pattern. KCNQs show a similar structure and function, and have peculiar regulatory properties, distinct from those of most other potassium channels.
GPV:What is the potential value of looking at all the Kv7 genes together synergistically in terms of helping understand KCNQ2 encephalopathy?
MT: At least in specific cell types and at distinct developmental stages, some members of the Kv7/KCNQ family, namely KCNQ3 and KCNQ5, are found in the brain, where they can work together with KCNQ2 and modulate its function. Mutations in KCNQ3 and, more recently, KCNQ5 have been shown to cause neurological diseases with features partially-overlapping KCNQ2 encephalopathy, although gene- and even mutation-dependent phenotypes are also emerging. Therefore, understanding structural and functional similarities and differences among KCNQ family members may potentially provide new strategies for the treatment of KCNQ2 encephalopathy; in fact, available Kv7-acting drugs all have serious limitations, including poor selectivity among Kv7 family members. For instance, information on specific pharmacological properties of cardiac Kv7.1 channels might facilitate the development of “brain” Kv7.2-specific drugs without the potential negative side effects that cause arrhythmias.
GPV:Conversely, how have advances in KCNQ2 research improved knowledge in the study of other KCNQ illnesses?
MT: As said before, despite the previously-mentioned similarities in functional characteristics and structure among members of Kv7/KCNQ family, each of them plays a different functional role at distinct sites in the human body; thus, the knowledge generated from KCNQ2 studies could provide the opportunity to target diseases completely distinct from KCNQ2-related diseases, such as hypertension, urinary dysfunction, pain, arrhythmias, and GI disorders.
GPV: What are your hopes for this conference?
MT: The goal of this conference is to facilitate the sharing of knowledge and ideas among scientists working in different fields in which the Kv7 channels play a relevant functional role; we hope that such cross-fertilizing experience will deepen our understanding of KCNQ-related illnesses and reveal emerging and yet under-explored areas in which Kv7 channels are involved, such as (just to mention a few …) cancer, depression, anxiety, and autism-related disorders. The meeting will also strengthen existing collaborative research networks and create new ones, further enhancing the opportunity for young scientists to train in prestigious labs around the world. Furthermore, given the participation of several pharmaceutical companies, the meeting will provide a unique opportunity to share results between academy and industry to speed the pace toward finding a cure for KCNQ-related illnesses.
Dr. Millichap is an attending physician in the Neurology and Epilepsy Center at the Ann and Robert H. Lurie Children’s Hospital in Chicago and Assistant Professor of Pediatrics and Neurology, Northwestern University Feinberg School of Medicine. The Jack Pribaz Foundation chose Dr. Millichap to receive the award for his dedication to bridging bench science and clinical application and for his contributions to the medical literature.
“At the heart of any pediatric medical research there is always a family who needs a champion to fight for their child,” said Mike Pribaz, President of the Foundation and one of the directors of the Board. “We are proud to honor Dr. Millichap with this award for his efforts to identify new patients with KCNQ2 mutations and to educate the medical community throughout the world about this rare condition. Dr. Millichap is a champion for the entire KCNQ2 family and the larger community of pediatric neurology.”
Liz Pribaz presents Dr. John Millichap with the first ever Sophie Award for Young KCNQ2 Investigators on January 23, 2016, at Lurie Childrens’ Hospital in Chicago. Mike Pribaz and son Jack hand off the big check.
“I accept this award with pride and a sense of excitement,” said Dr. Millichap. “It’s amazing to see how far we’ve come in such a short time. There’s tremendous momentum in this field of study and a vibrant international community of researchers and patient advocates to work with.”
A huge thank you to all the JPF donors whose generosity makes such moments–and such important work in KCNQ2 and rare epilepsy research–possible. We are truly Together in Search for a Cure!
Sophie was born in Arkansas to parents Sasha and Colten on January 4th, 2015. When Sophie was diagnosed with KCNQ2 at 4 weeks old, her parents did what most parents faced with this diagnosis do: whatever they can to help their children.
Sadly, little Sophie, who had further medical complications, died at just 13 months old. For Sasha and Colten, that desire to do whatever they could to help Sophie turned into a desire to help other KCNQ2 children. They made a tough decision and donated her brain to science. This donation will help in the study of epilepsy and KCNQ2.
So to honor Sophie and her parents’ tremendous gift, The Jack Pribaz Foundation is establishing a new award for KCNQ2 research. We are calling it the Sophie Award for Young KCNQ2 Investigators.
This is an award that looks bravely forward, like Sasha and Colten have. It is a grant in the amount of $15,000, designed to support and encourage research of KCNQ2-related epilepsy.
We are thrilled to announce the first Sophie Award winner today at the 70th Annual Meeting of the American Epilepsy Society in Houston, Texas. The recipient is Dr. John Millichap, attending physician at the Neurology and Epilepsy Center at the Ann and Robert H. Lurie Children’s Hospital in Chicago and Assistant Professor of Pediatrics and Neurology, Northwestern University Feinberg School of Medicine. We chose Dr. Millichap for his outstanding dedication to bridging bench science and clinical application and for his contributions to the medical literature. His work is advancing understanding of KCNQ2-related epilepsy into the next generation of scientists and clinicians.
Dr. John Millichap
“I’m so proud to receive this award and to be a part of KCNQ2-related epilepsy research,” said Dr. Millichap. “The spirit of collaboration I’ve found on an international scale among both researchers and families is uncommon and powerful. Together we have come a long way in a relatively short time, but there is a lot of work ahead of us,” he said. “Receiving the Sophie Award is exciting but also humbling, because at the heart of our efforts are children and families who desperately need science to catch up with their conditions.”
We are very passionate about this prize because it is a special opportunity to support KCNQ2 research and also to recognize the important partnership that KCNQ2 families have with researchers. That’s what Sophie’s gift was all about.
Look into the future with us. Gifts made to the Sophie Award for Young KCNQ2 Investigators will go to sponsor scientists, grad students, doctoral candidates, and young professors of the future in Sophie’s memory.
We sincerely thank Sasha and Colten for their dedication to the KCNQ2 cause. And we congratulate them, too– they had a new baby boy this summer! They give us so much hope!
Wouldn’t it be great to better understand the relationship between daily activity, sleep, emotional state, medication adherence, and epilepsy symptoms? Empatica’s new smartwatch, the Embrace, may be a useful tool in connecting those dots.
In our most recent issue of the Q2 News, Dr. John Millichap discussed the scientific background of the new technology. Read all about it here. The Embrace Alert System (Embrace watch and Alert app) detects and alerts to convulsive seizures, and a new clinical trial on the long-term effectiveness of this system will soon be underway.
Empatica announced approval of their clinical trial “Embrace Seizure Characterization” on May 11, 2016.
Hundreds of people received a free Embrace smartwatch thanks to the Epilepsy Foundation’s sponsorship and a give-one-get-one campaign. Kurt Florian, President and CEO of the Epilepsy Foundation of Greater Chicago, tells us that his organization helped about 50 families from Chicagoland apply for a free Embrace watch. My nephew Jack is in line to get one of them (thanks, EFGC!). The Epilepsy Foundation stopped taking applications March 27 of this year.
So if you still want an Embrace smartwatch, how can you get one?
To answer practical questions like this, I called on Empatica’s own David Bender, our “booth neighbor” for five days at the AES conference in Philadelphia. He was kind enough to answer some questions for us:
If a family missed the chance to get a free watch, how can they still get one?
The watch is still only available for pre-order at this time. Our production schedule is still ramping up to meet existing demand which we’re hoping to do by September 2016. We have validated the hardware design in preliminary usability studies this Spring with several hundred families and adults and were able to move ahead with the mass-production phase, meaning that the devices are now starting to ship.
Will there be any future opportunities to apply for a free Embrace?
We are incredibly grateful for the initial support of our Indiegogo crowd funders who sponsored give-one-get-one packages, which lead to 1540 donated devices that are being distributed in collaboration with the Epilepsy Foundation. At the moment our focus is on meeting the release schedules, and we do not have any current sponsors for free Embraces. We anticipate that once all back-orders are shipped and Embrace is in stock we’ll be in a good position to support more sponsorship programs. We’re also trying to keep the price of the system low (under $200) which we hope is affordable for most families until insurance reimbursement models are in place.
How can families enroll in the clinical trial?
Any individual who is scheduled to receive an Embrace watch will receive an invitation to enroll in the clinical trial through their Empatica Account. The enrollment process involves a screening (for example, we’re only enrolling US residents who are fluent in English and are not pregnant or planning to become pregnant at this time) as well as an informed consent review which families should review and ask questions about if there is anything unclear. Once informed consent is established the family can install the Empatica Alert app. Folks who do not wish to enroll can still use Embrace without the Alert app for the quantified self functionality (Empatica Mate app has sleep, autonomic stress, and activity reports, med reminders, and can be used as a context-rich seizure diary). For folks who are outside the US we’re working on a separate regulatory strategy for each region. We will make more announcements in the future so please stay tuned! For more info on the US trial please see our Embrace support pages: https://support.empatica.com/hc/en-us/categories/200310325-Embrace-watch
You have heard of Potiga, a drug used to treat KCNQ2-related epilepsy. But what about new drugs in development like SF0034 or RL648_81? KCNQ2 Perspectives asked renowned pharmacologist and Jack Pribaz Foundation scientific adviser Dr. Maurizio Taglialatela to provide an overview of potassium channel openers. We are grateful for his help in understanding these drugs and offering a glimpse of what promising new treatments may lie on the horizon.
Precision medicine is an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle. For genetic diseases such as most epileptic encephalopathies (EE), precision medicine aims to provide individually tailored treatments based the underlying genetic cause.
For KCNQ2-EE, molecular diagnosis may influence the choice of antiepileptic drugs. In fact, most of the genetic variants identified in KCNQ2-EE, when studied in vitro by conventional electrophysiological methods, cause a decrease in KCNQ2 channel function (loss-of-function or LOF variants). An antiepileptic drug which enhances KCNQ2 channel function may represent a novel therapeutic approach in those patients carrying LOF KCNQ2 variants.
Retigabine
Among currently available antiepileptic drugs, retigabine or ezogabine (trade name Trobalt or Potiga) is the only one whose clinical efficacy is related to this specific mechanism of action. Unfortunately, although retigabine is approved for use in adults with epilepsy, its use in infants and children is very limited. Moreover, post-marketing studies revealed the occurrence of side effects with retigabine, including a blue discoloration of the skin and mucosae and pigment deposition in the retina. It is yet unknown whether this latter effect, which appears as a consequence of the drug’s photodegradation and oxidation upon exposure to UV radiations, affects vision in the long term. Another side effect occurring with retigabine is urinary retention, possibly a consequence of the poor selectivity of retigabine/ezogabine for KCNQ2 channels over channels of the same subfamily (particularly KCNQ4), which do not appear to play a significant role in epilepsy, but which are involved in the control of bladder smooth muscle contractility.
SF0034
To improve retigabine safety and selectivity, in the last few years, several research groups have synthesized and characterized the pharmacological properties of a large number of retigabine analogs (as well as other KCNQ2 activators structurally unrelated to retigabine). Among the novel molecules resulting from these efforts is SF0034, a compound which, when compared to retigabine, is more potent and KCNQ2-selective (Kalappa et al., 2015). Preclinical studies in animals also revealed that SF0034 exhibits a more potent anticonvulsant activity and less toxicity than retigabine. In addition, SF0034 was significantly less active on KCNQ4 channels, leading to the suggestion that, in comparison with retigabine, side effects associated with activation of this channel subtype may be less likely to occur with SF0034. SF0034 seems to be more chemically stable and does not produce blue metabolites, suggesting that it may be a useful clinical candidate for epilepsy and other diseases linked to KCNQ2 hypofunction (such as tinnitus).
RL648_81
Even more recently, another analogue of retigabine, named RL648_81, has been reported in the literature (Kumar et al., 2016); this compound is 3 times more potent than SF0034 and 15 times more potent than retigabine in activating KCNQ2 channels in in vitro electrophysiological studies. The same studies also revealed that RL648_81 is also more selective than retigabine for activation of KCNQ2 channel over channels formed by other KCNQ subtypes. However, no pharmacokinetic data on drug absorption, metabolism, distribution and elimination from the organism is yet available for RL648_81 (either in experimental animals or in humans), and no antiepileptic efficacy has been proven for this drug in preclinical or clinical studies. Nonetheless, it seems likely that this more potent, selective and chemically stable KCNQ2/3 activator may be yet another attractive candidate for treating or preventing hyperexcitability disorders, overcoming some of the undesirable side effects associated with retigabine.
In conclusion, based on the discovery that KCNQ2 channels play critical roles in epilepsy pathogenesis and treatment, both academic and industry efforts are being actively pursued to develop novel KCNQ2 activators. Whether these novel drugs will prove useful as targeted therapies to improve neonatal seizures and developmental outcome in neonates with KCNQ2-EE remains to be demonstrated in the years to come.
Born in Naples, Italy, Dr. Maurizio Taglialatela, MD, PhD, is a full professor of pharmacology at the University of Molise in Italy, where he was Dean of the Faculty of Health Science from 2005-2012. He earned both his Medical Degree and PhD in Cellular and Molecular Biology and Pathology from the University of Naples. At the University of Molise he serves as the scientific director of the Center for Pharmacovigilance of the Regione Molise, the chairman of the Center for Higher Education of Public and Private Management of the University of Molise, and the coordinator of the PhD Program in Translational and Clinical Medicine. Dr. Taglialatela has contributed to numerous international research committees for pharmaceutical drug development. In the last fifteen years, he has dedicated most of his efforts to investigate the molecular pathogenetic mechanisms responsible for neuronal channelopathies, concentrating on developing experimental tools and drugs to investigate the structure-function analysis of disease-causing human mutations found in human KCNQ channel genes, in order to indentify in-vitro biomarkers useful for disease diagnosis, prognosis, and therapy.
Warm thanks to Tanya and John for sending this update to their son Oliver’s story, which first appeared many moons ago on the original Jack’s Army website and which now resides in the KCNQ2.org Stories section. It is so helpful to be able to revisit the “vintage” stories, celebrate progress, and learn from challenges. Sharing stories is a powerful way the entire KCNQ2 community can lift one another up and enlighten researchers and clinicians about the daily realities of living with KCNQ2-related epilepsy.
Our sweet little Oliver is now four years old, two and a half years after diagnosis of KCNQ2 at the age of 17 months. On the whole, Oliver is non-verbal; however, he teaches us how to communicate with him on a daily basis, such as using his eyes and his hands when he wants something!
Physically he is doing an awesome job. He can run, jump, and climb. However, he does tend to walk on all fours, which is a typical autistic trait.
Currently he attends a special school with two to three other children in his class. He goes there one day per week. He also attends a mainstream Kindy five days a fortnight. There he has his own special needs assistant looking after him. He certainly keeps her on her toes! The children have been taught about autism and his epilepsy. This was brought on by Oliver and his love for opening doors and gates. His way of communicating is taking your hand (any hand, even a complete stranger’s!) and leading you to where and what he wants. Because this was becoming an issue, we sent out letters to other parents in the school group (about 20 in all) about autism and KCNQ2. We have been given positive feedback. One person has even asked about more information, which is very exciting.
“Our KCNQ2 community is built on friendship, common goals, and empathy.” –TG and JG
Since Oliver’s diagnosis, things have been on the move. Sometimes up, sometimes down. On the seizure front, he has had 3 seizures in the past 3 years. Not bad, but one seizure is one too many. We were weaning his main AED [anti-epileptic drug] just recently. After two days of completely being off it, he had a seven-minute seizure. After advice from his neurologist, we restarted the medicine. That has been disappointing as we wanted to see what his personality and cognitive development would be affected without any.
He is also been receiving additional therapies such as speech and occupational therapy. They are slowly making inroads with his communication skills.
Oliver is also doing great with his personal care. A couple of years ago, the thought of having his hair washed was exhausting. Now he loves the water and having his hair washed is a lot easier! At times he can help getting dressed. We need to help him a lot, but it is a step in the right direction.
I think parents need to listen to other parents about KCNQ2. Tell us your story. Every time I tell Oliver’s story to friends, doctors, etc, I feel as though I am educating them through our experiences. Unfortunately there isn’t a lot known about it in general medical circles. Education is all about experiences. For example, when Oliver was brought in to the emergency department after his last seizure, an emergency consultant asked what it was! Unfortunately, this is a reoccurring theme.
The other thing I would suggest is collaboration is the key. Talk to all of your child’s healthcare providers so they can collaborate the best that they can for your child.
The most important thing is to hug and love your child. Our KCNQ2 community is built on friendship, common goals, and empathy. We are the only ones in the world that exactly know what you are going through. Never be afraid to ask or share. We are here for our children and our community.
Oliver was born in February 2012 through an elective caesarean section. On day two of life, while he was breast feeding, Oliver broke attachment and started staring and all four limbs were twitching. The next day, he had two tonic clonic seizures lasting about 45 secs each time. He was then transferred into the hospital’s special care nursery. There he had numerous tests including a lumbar puncture, MRI, ultrasounds and many blood tests. All of these tests came back normal. A week later, he was transferred to a larger hospital for extensive and specialised testing, including a few EEG’s.
Over the next couple of months he had numerous hospital admissions due to increase seizure activity and the inability of his medications to keep up with them. After his scheduled four-month vaccinations, he had his longest seizures to date. A seven-minute seizure at home, an eight-minute seizure at hospital, then had a 45-minute status and transferred to the Intensive Care Unit. It was after that enormous seizure that his neurologist decided to do genetic testing. After a couple of days, he was fine and sent home.
Over the next four to six months, his hospital admissions due to seizure activity were getting less. However, he was not putting on any weight, his appetite decreased, and it got to a stage where his output was getting less and less. His doctor decided to do some blood tests. His electrolytes were at a critical level and with consultation with a metabolic doctor, decided it was his current anti-epileptic drug was the root cause of this. We changed medications immediately.
In July 2013, we discovered that Oliver was diagnosed with the KCNQ2 gene mutation, the cause of his epilepsy. Through the internet, we have discovered many families that are affected by this and have been a tower of strength for all of us.
At 22 months of age, he had been seizure free for 10 months and is physically growing well. Developmentally he is quite behind children of a similar age. He started walking at 19 months of age. Still quite non-verbal, and it is like he is in his own little world. He has had a recent diagnosis of autism; fortunately, he is on the lower end of the spectrum. He receives regular physical therapies through occupational therapists, physiotherapists and speech pathologists. Because there isn’t much known about KCNQ2, the doctors are learning as we are learning. We continue to support his needs and whatever he requires. We take it day by day and try to live life as best we can.
Share your KCNQ2 journey here to help raise awareness of KCNQ2-related epilepsy.
Boston genetic counselor Lacey Smith, MS, CGC, shares the most common questions she hears from families.
I have been a genetic counselor in the Epilepsy Genetics Program at Boston Children’s Hospital for about three years. During my time here, I have met with many families who have received a genetic diagnosis for their child’s epilepsy, including KCNQ2-related epilepsy. Getting such a diagnosis can be overwhelming for families, as there is often a lot of information coming in at once. My hope is to provide you with answers to some of the questions I am most often asked by families when they first hear that their child has KCNQ2-related epilepsy. Even though your child has this diagnosis, each individual is unique so your experiences may be different from those of other families you may have met who also have a child with KCNQ2-related epilepsy.
The spectrum of features associated with KCNQ2-related epilepsy is quite vast and the genetics aspect is often unique to each family, so my answers may be a little vague and may not touch upon your specific needs or experiences. As such, I am also providing suggestions of whom to contact to gather additional information specific to your child. You can also use this information as a guide in coming up with questions to ask your child’s neurologist.
What does a diagnosis of “KCNQ2-related epilepsy” mean?
This means that your child has an alteration (also called a “variant”) in a gene called KCNQ2, and that variant has likely caused your child to develop seizures or epilepsy. It may also explain any developmental challenges your child may have.
Knowing the underlying genetic cause of your child’s epilepsy could be beneficial in a number of ways. First of all, it provides an answer or a reason as to why your child developed seizures. Having a diagnosis can allow your child’s doctor to provide you with more information regarding prognosis and potential treatment options than they would have been able to without knowing the underlying cause. It can prevent your child from undergoing additional unnecessary tests to rule out other conditions or medical complications. It can provide insight into family history if there are any other family members who have/had seizures and it can provide information regarding the likelihood of having another child with similar symptoms.
What does this diagnosis mean for my child’s prognosis?
Knowing that your child has a variation in the KCNQ2 gene can allow your child’s doctor to have a better idea of how your child’s symptoms may progress over time, based on what is already known about other children who have KCNQ2-related epilepsy. There are two general categories of symptoms. Children usually fall somewhere along a spectrum from Benign Familial Neonatal Epilepsy (BFNE) on the mild end to an epileptic encephalopathy. In the world of epilepsy, “benign” usually means that seizures are expected to resolve over time or the child may “grow out” of their seizures. An epileptic encephalopathy is a term that is used when a child has seizures, and in addition, the electrical activity of the brain both during and between seizures can also cause developmental delays and cognitive challenges.
So how does one know where along the spectrum their child will be? Unfortunately, this is difficult to know. Currently, this isn’t enough information available to predict exactly what symptoms a child will develop based solely on the results of the genetic test. Even among individuals who have the same KCNQ2 variant, each individual can have different symptoms. There are likely other factors (such as other genes) that are modifying the effects of the KCNQ2 variation. Researchers are trying to learn more about these contributing factors. In the meantime, your child is going to tell his or her own story. Most often, the best predictor for how your child will do is how your child has done/is doing. This can be difficult when the diagnosis comes at a very young age and there is little developmental history to look back on.
…your child is going to tell his or her own story. –LS
Over time, researchers will hopefully gain a better understanding of features associated KCNQ2-related epilepsy. By gathering information from many individuals who have been diagnosed, they can begin to analyze patterns including the spectrum of symptoms and look for any associated modifiers to be able to better predict a child’s prognosis. One of the ways in which researchers can collect this information is through a patient registry. A registry gathers and keeps information about people who have a certain condition to support and encourage research into that condition. Individuals may sign up or enroll in a patient registry to have their information included and available to researchers. The RIKEE Project is one patient registry that is targeted specifically to KCNQ2-related epilepsy.
Register your child in RIKEE to connect with the international team of KCNQ2 researchers and advance understanding of KCNQ2-related epilepsy.
How does this diagnosis affect management/treatment for my child?
This diagnosis may not lead to a drastic change in management. For example, if your child has been experiencing some delays in development and has been receiving services to help with development, then he or she would continue to receive those services. If your child is on target for all milestones, then you will know to be on the lookout for any delays moving forward and intervene right away with evaluations and/or services if needed.
You may find that your child’s doctor may not recommend any medication changes based on this diagnosis, especially if your child’s seizures are well-controlled. Targeted treatments for genetic forms of epilepsy is a very big topic in research right now– being able to treat the exact cause of seizures based on the underlying genetic cause is on the horizon and new advancements are happening quite frequently. This is true for KCNQ2-related epilepsy. Because the field is evolving so rapidly, it is best to discuss medication management with your child’s doctor in order to have the most up-to- date information.
If this is “genetic,” does it mean that one of us parents passed this on to our child?
Not necessarily. Just because a condition is considered “genetic” does not necessarily mean it was inherited. Genetic variations can be passed through families, or these variations can arise for the first time in a child (these are called “de novo” variants). De novo variants are likely produced by a spontaneous event during the formation of the sperm or the egg cell. Some individuals can have the KCNQ2 variant but never develop any symptoms, so it is possible for a child to inherit the variant from a seemingly unaffected parent. You can determine whether your child has an inherited variant or a de novo variant by testing both parents. Your child’s doctor can help facilitate such testing.
What does this mean for my other child/children? Can I test them?
If you have another child who also has seizures, then it is possible that they could also have the same KCNQ2 variant and it would be possible for this child to have genetic testing. In general, it is not routinely recommended that unaffected children (children who do not show any features of the condition) undergo testing. Rather, that child could make the decision to undergo testing on their own when they are of suitable age.
What are the chances of having another child with this condition? Can I reduce the chances?
The likelihood of having another child that has the KCNQ2 variant depends on whether this was an inherited variant or a de novo variant. If the KCNQ2 variant was inherited from a parent, then there would be a 50% (1 in 2) chance that each conception would also inherit the KCNQ2 variant. If the KCNQ2 variant was de novo, then the chances are much smaller although not zero. This is because there is some residual chance to have another child with the same variant due to something called “gonadal mosaicism.” Gonadal mosaicism is not something that is routinely tested for in the clinical setting. For some families, during the parental testing process, the KCNQ2 variant is detected in some, but not all, of the cells in one of the parents. This is another form of mosaicism. For those families, it is also possible for future pregnancies to also have the KCNQ2 variant.
Regardless of inheritance of the variant it is possible to test future pregnancies to see if the KCNQ2 variant is present. For more information regarding prenatal testing options, you can speak with a genetics specialist when you are considering having additional children.
It is also important to remember that individuals with KCNQ2 variants or KCNQ2-related epilepsy may show different symptoms, even when they have the same variant. While it is possible to test pregnancies for the KCNQ2 variant, one cannot predict with certainty the severity of symptoms an individual may present with. Future children who have the KCNQ2 variant may have symptoms that are less severe or more severe than those of your child.
Who else can I talk to?
It is important to find a neurologist or epileptologist with whom you/your child feel comfortable. Don’t be afraid to ask for a second opinion, or more! If you have concerns regarding your child’s development, you could have your child evaluated by a neuropsychologist. A neuropsychologist is a doctor who, through a series of tests, can identify your child’s strengths and weaknesses in learning, emotions and behavior. From there, recommendations for services specific to your child’s needs can be made.
It may be helpful to speak with someone who has specific training with genetic conditions, whether it be specifically genetic forms of epilepsy or more general genetic conditions. This could be a geneticist and/or a genetic counselor. Healthcare providers who are specially trained in genetics can provide information regarding inheritance patterns, identifying family members who could be tested and options for testing future pregnancies, if desired. You may consider asking your child’s doctor to refer you to a genetics specialist in your area. You or your child’s provider can also find a genetic counselor through the National Society of Genetic Counselors website at www.NSGC.org, and navigate to the “Find a Genetic Counselor” link. You can identify genetic counselors in your area and genetic counselors that specialize in epilepsy or neurogenetics.
Lacey Smith, MS, CGC
Lacey Smith, MS, CGC, is a genetic counselor in the Epilepsy Genetics Program at Boston Children’s Hospital. She has roles in both the clinic and in research. In the clinic, she works closely with epileptologists in meeting with children who either have, or are presumed to have, a genetic form of epilepsy. She reviews all aspects of genetic testing, discusses potential implications for other family members and future children, and helps families understand their child’s diagnosis. In the research world, her team is trying to better understand the genetic contributions to epilepsy. Specifically, they are trying to identify new genes associated with epilepsy, determine how variations in these genes cause epilepsy, and develop more targeted therapies for children who have epilepsy caused by variations in these genes. Lacey is also a member of EpiGC, a consortium of genetic counselors whose mission is to promote access to quality genetics services for patients and families affected by epilepsy through research, public policy/advocacy, education and outreach activities.
By all accounts, KCNQ2 is getting serious attention. On New Year’s Eve 2015, Dr. Ingo Helbig, one of the authors of the blog Beyond the Ion Channel, recapped his top ten posts for the year (hopefully while wearing a party hat and sipping a little bubbly). KCNQ2 came in at #10, and he wrote that KCNQ2 “may be the most common genetic cause of neonatal epileptic encephalopathies.” For any family who has ever felt alone with a KCNQ2 diagnosis, that’s a pretty incredible thing to read. It spurred me to ask for Dr. Helbig’s perspective for KCNQ2 Perspectives, and he has graciously obliged.
You have invented this new term, Epilepsiome. Please tell us what the Epilepsiome is and how researchers and clinicians can use it.
The Epilepsiome is the community-based gene curation effort of the epilepsy world. Basically, the Epilepsiome project aims to review and curate data on all relevant epilepsy genes, especially involving scientists and clinicians who work on the particular genes. The goal is to provide you with an up-to-date overview that will then link out to the other resource such as ClinGen, ClinVar, GeneReviews, or OMIM. However, in contrast to these resources (which are used widely in genetic studies), the Epilepsiome will have a clear focus on epilepsy and will put the current findings and controversies into context. My vision was to carry some blogging atmosphere over to a gene curation project, which typically engages the community more than a static page that is only updated infrequently.
The Epilepsiome is growing. We are building expert groups for particular genes and we are very fortunate to have a dedicated group of scientists and clinicians on board to add to our gene curation. We aim to have the main genes for genetic epilepsies reviewed in 2016–and yes, there is already a KCNQ2 page maintained by some of the experts in the field that are part of your community.
In your New Year’s Eve 2015 post you wrote: “In many ways, KCNQ2 is one of our model genetic diseases, with a tightly connected group of researchers, clinicians, and patient organization. We hope that we can spin this energy into novel approaches for precision medicine soon.” Can you tell our readers what you mean by precision medicine and how the KCNQ2 community might be able to contribute to, or benefit from, it?
I knew that you would pick up on this! I must say that I am really impressed by the KCNQ2 community, which connects families, clinicians, and researchers. The vision of precision medicine is to use our knowledge of the genetic causes to find custom treatments, which may either be known medications that we did not think of yet or new compounds that were designed for specific disorders.
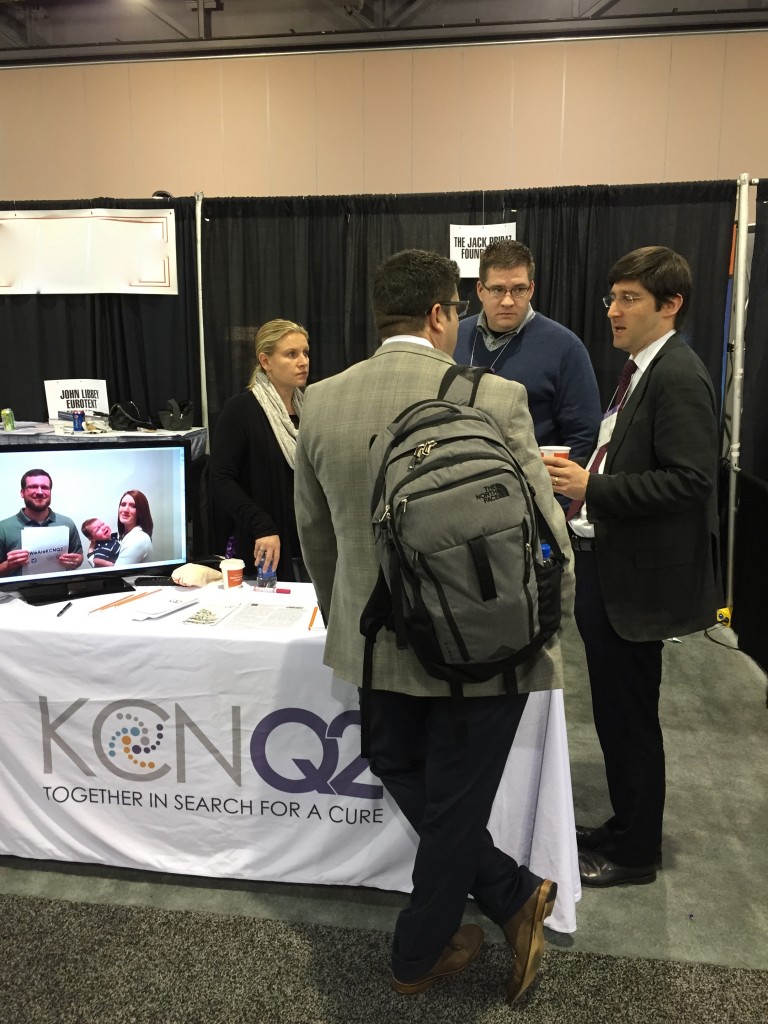
JPF booth at AES in Philadelphia. KCNQ2 families sent in #WeAreKCNQ2 videos to demonstrate their spirit and unity to the research community. Pictured: in video, baby Parker and family; Liz Pribaz, Jim Thompson, Dr. John Millichap. View the #WeAreKCNQ2 video on our YouTube channel.
We feel that this approach may be promising as we are facing two fundamental problems in many severe epilepsies. First, seizures in these epilepsies are often difficult to treat and secondly, other than controlling seizures, our treatment strategies often have little impact on the developmental outcome. Precision medicine or personalized medicine aims to identify the cause of the epilepsy first. For patients with KCNQ2 encephalopathy, this is often a mutation that renders the potassium channel non-functional. The idea of precision medicine is to find model systems that replicate this defect and then systematically search for compounds that may correct it. A closely-knit community as the KCNQ2 community will be required to make sure that future treatment options can quickly be translated to clinical trials. Many people underestimate the efforts of finding patients with rare diseases for trials, and I feel that having the community organized is a prerequisite for this.
A closely-knit community as the KCNQ2 community will be required to make sure that future treatment options can quickly be translated to clinical trials.–IH
For parents of children suffering seizures and other effects of a genetic mutation, no science can ever move fast enough to find a cure or better treatment. What does that timeline for precision medicine advances look like?
Unfortunately, it is difficult to provide timelines for this. In many aspects, we are still not at the stage yet where generating model systems and testing of a wide range of possible candidate drugs is a predictable pipeline. Having said this, there is considerable effort dedicated towards finding therapies for patients with ion channel diseases.
There is one additional aspect to treatment of genetic epilepsies that I would like to mention at this point. You mentioned the word “cure.” While both families and clinicians often expect the scientific community to come up with game-changing interventions that significantly alter the course of the disease, a different strategy is to systematically focus on exploiting small gains in the field.
Take the pediatric leukemia field as an example. Even though many individual chemotherapy and radiation therapies have increased survival, it was only the thoughtful combination of therapies and systematic adaptation of protocols based on prior experience that has led to the significant increase in cure rates that we see today. Translating this to genetic epilepsies, I feel that we already can learn a lot from existing experience, but access to that data, how best to aggregate it while protecting patient privacy, and the possibility to mine this information are major issues.
Until recently, public discussion of scientific ideas was the purview of peer reviewed articles and books. The internet, and especially blogs like Beyond the Ion Channel, have created new space for sharing ideas and debate. As a scientist-blogger, what do you see as the benefits and risks of these new public platforms?
Basically, these public platforms serve as the communication backbone of communities that are not represented in the traditional publishing format. The main benefit of these platforms is that they provide a “running commentary” on what is happening in the area; they help put novel and existing information into context. I have always felt that we are piecing together the puzzle of an ongoing mystery, the genetic architecture of a disease. And we are trying not only to look at whether the pieces fit, but also to present the bigger picture on what they contribute. Another big advantage of these platforms is the fact that they are updated regularly in small increments as they follow the rhythm of social media. This is different to the traditional format where only the final product is presented. Through these platforms, we are able to follow the thought process and generation of ideas, which gives us the possibility to interact and contribute. Finally, I have also felt the effects of blogging personally. It is a way for me to clear my mind and experiment with novel ideas. It’s the thing that keeps me up at 3AM.
With regards to risks, especially for online platforms that develop a certain authority in a healthcare-related area such as the rare disease space, you have to be very careful that the information on treatment and prognosis that you provide is solid and agreed upon by the community. We are currently in the process of adding a “Personalized Medicine” subsection for our gene reviews and have the discussion in the community on what constitutes reliable information that can be displayed in this section. This is particularly difficult given the publication lag in this field–there is much knowledge and experience in the community that is unpublished and representing this adequately remains a challenge.
How did you first become interested in the study of genetic epilepsies?
During my medical doctoral thesis in Heidelberg, Germany, I worked on neuronal gap junctions or “electrical synapses,” direct cell-to-cell connections between neurons. This type of communication between nerve cells had been forgotten for a long time, but was rediscovered in the early 2000s, and we characterized the proteins that were involved. At one point, we speculated that mice deficient in one of the critical proteins called Connexin 36 may have a reduced threshold for seizures, as this protein was thought to enhance the connection between so-called inhibitory cells (neurons that are typically involved in reducing the activity of other neurons).
We could not validate this hypothesis and actually found the opposite. We also tried to assess whether variants in this gene may predispose to epilepsy, but there was nothing to be found. Independent of my disappointment, this started my interest in genes that predispose to epilepsy and also left me with a fundamental skepticism of falling in love with any particular hypothesis too much. On our blog, I am often referring to the power of team science and hypothesis-free approaches in genetics, which is somewhat a reference to my earlier disappointments.
Independent of my disappointment, this started my interest in genes that predispose to epilepsy and also left me with a fundamental skepticism of falling in love with any particular hypothesis too much.–IH
My other “key moment” happened during a trip to Australia when I interviewed for positions in various fields, some of which did not include epilepsy at all. As I mentioned in a post last year, I was sitting in a rock pool at Red Johanna Beach in Victoria, Australia, reading Nigel Tan’s “The truth is out there,” a 2004 review on the state of association studies in epilepsy. The writing style and big picture view of his review made me realize that epilepsy genetics is something that I might actually be interested in. I typically publish a post “Red Johanna Day” every year, which I consider my birthday in epilepsy genetics.
These two stories were my early beginnings. In retrospect, I think it is interesting how my strong clinical interest in genetic epilepsies was born out of a primarily scientific interest. The clinical part was something that I learned and became fully invested in while getting involved in patient care. To me, being a clinician and a scientist is an interesting combination that allows me have both the care of the individual patient and the larger picture in mind.
Have you registered your child or patient in the KCNQ2 patient registry? Access RIKEE here to help the KCNQ2 community.
Sheila M. sent us the story of her son John, a three-year-old with an inherited KCNQ2 mutation. We thank her for sharing her family’s journey, which highlights how complex KCNQ2 epilepsy is—reflecting a complicated set of genetic and environmental factors that can result in different outcomes, even among family members with the same variant. By sharing stories and registering in the RIKEE patient registry, parents can equip researchers to better study these complexities.
John was born November 16, 2012. At 36 hours of life and at home (we had been released from the hospital) John began having tonic clonic seizures. He was in the NICU at the Montreal Children’s Hospital for a month and was released on four medications. At three months old he had more seizures that could not be controlled and was apneic. He was in the PICU for a month again, and released with four medications, with Phenobarbitol being one of them.
After that his seizures were less frequent, febrile in nature, and closely associated with his vaccinations, but were overall controlled by medication. We were diagnosed with Benign Neonatal Familial Epilepsy (BFNE) secondary to KCQN2 genetic mutation in August of 2013. I say we because my entire family had genetic testing and my mother, my brother, my son, and myself were identified with the mutation. My father and John’s father were not.
My mother and I both experienced seizures in infancy, but given the lack of medical information at that time, we were just told that we had seizures. Eventually my mom and I were lucky enough to stop having them after about two years old.
My brother continues to be medicated for seizures, although his last seizure was three years ago. There are no known cognitive deficits for him, my mother, or me that we are aware of.
I knew I had had seizures as in infant and was terrified I would have them during pregnancy, but I hadn’t assumed my son would have a seizure disorder. In retrospect I could have pursued genetic testing prior to becoming pregnant, but I never imagined KCNQ2 existed or had knowledge of the relationship between seizure disorders and genetic mutations. And my doctor was a high-risk pregnancy specialist.
Did you know? In a 2015 study, 27 of 33 families with BFNE had a KCNQ2 mutation. Other implicated genes in the studied population include KCNQ3 and SCN2A.
Searching for supportive services
John is three years old now, and since he turned 18 months old we have been on a journey of heartbreaking developmental issues. It has been a tough month as our private daycare just asked us to leave because it was time to switch groups and they feel John is too developmentally delayed to be integrated with the three-year-olds.
John at one year
At 18 months developmental delays began being investigated by doctors in the neonatology department. John has been identified with a severe speech delay, mild intellectual delay, and has a sensation seeking profile. He is also on a waiting list at the Autism clinic at the Montreal Children’s Hospital.
How are epilepsy and autism related, if at all? Two articles, here and here, describe the complexities and controversies of research in this area.
Privately we have spent our money on speech therapy since last January. Recently we have had a social worker through thecentre local de services communautaires, or local community service centre (CLSC) and an occupational therapist who works with our son weekly. In addition, we are set to begin to receive services at the Mackay Center weekly beginning in January.
My husband and I both have to work full time and we spent more than a month searching for childcare. We were even rejected by a renown special needs pre-school as our son was identified as non-compliant and unworkable. We finally found a private daycare that is costly, and on top of the regular fee they are insisting John does additional therapy at additional cost because of his compliance issues. Unfortunately we cannot be reimbursed by insurance for that as he has no diagnosis of autism (which we do not think he has).
As challenging as his medical issues were, what we are working with now is heartbreaking, but we love our son who is a sweet, affectionate, and kind boy.
Share your KCNQ2 journey here to help raise awareness of KCNQ2-related epilepsy.