Every year–for five years now–we’ve had so much fun raising money for KCNQ2 research and awareness at our annual golf outing. We have been blessed and energized by the company of other KCNQ2 families at this event, and this year we are excited to announce that we are expanding it. We want you to join us!
This July we will be hosting a family-focused weekend before our golf outing. It’s designed to bring families together in a fun, relaxing, informative, and inspiring format. Part retreat, part seminar, part think tank, the KCNQ2 Family Roundtablepromises to be your best long weekend getaway of the year! And if you like to golf, well that’s just icing on the cake.
Sophie was born in Arkansas to parents Sasha and Colten on January 4th, 2015. When Sophie was diagnosed with KCNQ2 at 4 weeks old, her parents did what most parents faced with this diagnosis do: whatever they can to help their children.
Sadly, little Sophie, who had further medical complications, died at just 13 months old. For Sasha and Colten, that desire to do whatever they could to help Sophie turned into a desire to help other KCNQ2 children. They made a tough decision and donated her brain to science. This donation will help in the study of epilepsy and KCNQ2.
So to honor Sophie and her parents’ tremendous gift, The Jack Pribaz Foundation is establishing a new award for KCNQ2 research. We are calling it the Sophie Award for Young KCNQ2 Investigators.
This is an award that looks bravely forward, like Sasha and Colten have. It is a grant in the amount of $15,000, designed to support and encourage research of KCNQ2-related epilepsy.
We are thrilled to announce the first Sophie Award winner today at the 70th Annual Meeting of the American Epilepsy Society in Houston, Texas. The recipient is Dr. John Millichap, attending physician at the Neurology and Epilepsy Center at the Ann and Robert H. Lurie Children’s Hospital in Chicago and Assistant Professor of Pediatrics and Neurology, Northwestern University Feinberg School of Medicine. We chose Dr. Millichap for his outstanding dedication to bridging bench science and clinical application and for his contributions to the medical literature. His work is advancing understanding of KCNQ2-related epilepsy into the next generation of scientists and clinicians.
Dr. John Millichap
“I’m so proud to receive this award and to be a part of KCNQ2-related epilepsy research,” said Dr. Millichap. “The spirit of collaboration I’ve found on an international scale among both researchers and families is uncommon and powerful. Together we have come a long way in a relatively short time, but there is a lot of work ahead of us,” he said. “Receiving the Sophie Award is exciting but also humbling, because at the heart of our efforts are children and families who desperately need science to catch up with their conditions.”
We are very passionate about this prize because it is a special opportunity to support KCNQ2 research and also to recognize the important partnership that KCNQ2 families have with researchers. That’s what Sophie’s gift was all about.
Look into the future with us. Gifts made to the Sophie Award for Young KCNQ2 Investigators will go to sponsor scientists, grad students, doctoral candidates, and young professors of the future in Sophie’s memory.
We sincerely thank Sasha and Colten for their dedication to the KCNQ2 cause. And we congratulate them, too– they had a new baby boy this summer! They give us so much hope!
It seems like families receive new diagnoses of KCNQ2 on a weekly basis, but some “old timers” can remember a time when there were only a handful known to each another. Gillian’s family first shared her story almost three years ago, and we want to thank mom Nancy and for this wonderful update. Sharing stories represents not only a special generosity, but also the very real need to connect and know we are not alone. That is as true now as it was when we first met this special young lady.
It has been three years since Gillian’s diagnosis. Today at the age of seven she continues to be an active and happy girl. She has finished the second grade and is doing very well in school. She loves listening to music and dancing. She enjoys swimming and gymnastics, especially jumping on the trampoline.
Life with Gillian is unpredictable. As an infant we parented Gillian much in the same way you would any other baby but with the added worry of managing her meds. As she began to grow her inconsistent sleep patterns resulted in severe temper tantrums. This combined with Gillian’s inability to communicate her needs made life difficult at times. Although she is still nonverbal her receptive communication is fairly good and she is better able to understand what we say. Her expressive language is also coming along and we have begun using an iPad to help her communicate.
Gillian’s behaviours are less frequent these days but physically more difficult to manage. We love our little girl and her determination and hard work continues to amaze us. We feel very blessed to have such and amazing community of KCNQ2 families with whom we can share both the good and difficult times. It would have been great to have their support early on as we have learned so much from them in the past three years. Gillian’s future continues to be uncertain but we no longer have to face it alone. Together our amazing KCNQ2 kids have so much to teach the world.
On December 29, 2008, we gave birth to a beautiful baby girl. Although she was initially quiet, her newborn cry soon filled the air. We were overcome with joy and love that we were eager to take her home to meet her big brother. After waiting the requisite 24 hours we took our beautiful baby girl home, unaware that anything was wrong. Friends and family dropped by and we celebrated the end of a very happy year.
By the morning of January 1 we were sure something must be wrong. Gillian was making odd movements, her body was stiffening, her head turned to the side, and her eyes deviated followed by a loud-pitched cry and rapid heart rate. A trip to our local children’s clinic confirmed she was having seizures. We took her to the Hospital for Sick Children in Toronto. While in triage, Gillian had another seizure and was immediately admitted and taken for testing. Blood tests, a spinal tap, CAT scan and a MRI all followed. With the exception of an EEG, which showed abnormal brain activity, all tests were negative. Gillian was admitted to the hospital, and over the next 28 days she would continue to have seizures. With the help of three different medications Gillian had her last seizure in hospital on the day we brought her home.
Over the next few years we parented Gillian without a diagnosis as genetic and metabolic testing continuously produced negative results. Despite obvious developmental delays, Gillian eventually learned to walk at age two. In the spring of 2011, at the age of 3½, Gillian was finally weaned off her last anti epileptic. Genetic testing continued to produce negative results and we were prepared for the possibility that we might never know what was truly wrong with our daughter.
It was not until a year later on request by her neurologist for one more genetic test that we received a positive diagnosis for a mutation on her KCNQ2 gene. Although we now had a name for her condition, doctors could not tell us anything new about her future and we felt once again alone on this journey, until we came across Jack’s Army and the work of Doctor Cooper. Suddenly our family began to grow to include so many amazing children and their parents. Today at the age of five Gillian is an active and happy girl with a slightly mischievous side who enjoys listening to music, dancing, running and playing. Although she cannot speak she lets us know everyday how much she loves us. We are constantly amazed by our little girl and looking forward to her future.
What does life with KCNQ2-related epilepsy look like?
To a researcher, it’s a miscoded ion channel in the brain. For a neonatal doctor or epileptologist, it might look like a chaotic EEG you must explain to anguished parents. Ask those parents what life with KCNQ2-related epilepsy looks like, and you’ll get a thousand answers–and even more questions.
But what does KCNQ2-related epilepsy look like through the eyes of a sibling?
“Owen and Rhys are as typical as any brothers can be,” says mom Natalie. “They love each other and are best friends.”
“I want to make people understand what this gene mutation is about. That’s what I care about,” says the first-time filmmaker. “I want people to feel—to know that these kids’ lives matter.”
Big night
Owen accomplished that goal last night just by participating in the event. But even more exciting, the film won the filmpossible award, plus a trophy and $500! His mom Natalie Boese says that after accepting his prize, Owen returned to his seat “very emotional and had tears of joy.”
“I’m happy!” says Owen. “I feel lucky that I won this award because I am helping out Jack’s Army and all of Rhys’ KCNQ2 army. I am going to keep making movies to spread more information about KCNQ2.”
Owen accepts his award on stage, with Rhys’s image beaming overhead.
“We would like to congratulate Owen McIntyre on winning the filmpossible award,” said Stewart Wong, vice president, communications, marketing and advocacy for Holland Bloorview. “Our Children’s Advisory Council selected the winner, and many of the members said they could relate to the sibling relationship and thought Owen did a great job providing insight into his and his family’s day-to-day life.”
When asked how she felt watching her boys last night, Natalie said, “Owen was up on stage at TIFF supporting his little brother in the best way he knew how. Rhys is always there pushing Owen to think differently and creates continued compassion, compassion in a way that may have never been explored had Rhys not been born.”
Toronto’s CBC’s Metro Morning radio show host interviewed the young producer/director and his mom.
For the assignment, the students had to research their topic and conduct an interview. Owen found www.KCNQ2.org and called to talk with Mike Pribaz, The Jack Pribaz Foundation (aka “Jack’s Army”) president and Jack’s dad. Mike remembers Owen’s “very young but very confident” voice on the phone last fall.
“Owen is exactly the kind of brother that KCNQ2 kids need,” Mike says. “He is thoughtful and invested in the future of his own brother as well as the future of all these kids. We need professionals and parents to spread awareness, but it is amazing to see such a bright young man do the same.”
Owen notices the looks that his brother gets from strangers. “When people see kids with disabilities their jaw drops when they don’t understand. I want people to understand,” he says. “I want people to smile normally.”
Anyone who watches Owen’s film can’t help but smile. It invites viewers to share a day of his life with a KCNQ2 sibling. While Rhys’s challenges are apparent, the film celebrates Rhys and radiates the love between brothers with tenderness and humor.
That’s exactly what the film festival is looking to accomplish. “Holland Bloorview Kids Rehabilitation Hospital is excited to partner with TIFF Kids International Film Festival this year to present our fifth annual filmpossible award, which recognizes young filmmakers who bring visibility to disability,” says Wong. “All of the filmpossible finalists this year did an amazing job giving us a window into their lives and helping us experience what they see and hear and feel every day. By sharing their joys and struggles, these very talented young filmmakers are helping to break down stereotypes and encourage compassion and understanding.”
“I want people to feel—to know that these kids’ lives matter.”–O.M.
Perhaps Owen gets his eye for beautiful images from his mom, who had this to say about her boys:
“We believed at first Rhys was at first our little turtle, slow and steady and coming along. As he grew up he became our unicorn, even compared to his KCNQ2 friends he is so unique– no one has the same gene sequence as he. He is as special and beautiful as they come. Both of my boys make me proud every day because of the way they view life, not so much in the awards and milestones they achieve.”
Specialness of siblings
Brothers sharing a good laugh.
Owen believes that being Rhys’ sibling puts him in a unique position, in some ways closer to Rhys than anyone else. “Parents don’t understand as much as siblings do,” Owen says. “Siblings have a connection.” He explained that being Rhys’s brother means he naturally treats him as an equal.
Although he admits they don’t quite fight like brothers usually do (“the gene mutation sort of pauses that,” he explains), he knows that his brother is engaged with him and has opinions and preferences all his own. Owen says Rhys definitely “has something to say back!”
Wrestling with big questions
Owen faces down some profound questions when he thinks about his brother’s future. He understands that science takes time, maybe a long time, to come up with the kind of treatment that will help Rhys. He’s “not so happy” about that.
But he also knows that there are people helping. And he wants to be one of them. He hopes his film will move people to learn more about KCNQ2. “You’ve got to care,” he says. “You’ve got to feel it.”
Sheila M. sent us the story of her son John, a three-year-old with an inherited KCNQ2 mutation. We thank her for sharing her family’s journey, which highlights how complex KCNQ2 epilepsy is—reflecting a complicated set of genetic and environmental factors that can result in different outcomes, even among family members with the same variant. By sharing stories and registering in the RIKEE patient registry, parents can equip researchers to better study these complexities.
John was born November 16, 2012. At 36 hours of life and at home (we had been released from the hospital) John began having tonic clonic seizures. He was in the NICU at the Montreal Children’s Hospital for a month and was released on four medications. At three months old he had more seizures that could not be controlled and was apneic. He was in the PICU for a month again, and released with four medications, with Phenobarbitol being one of them.
After that his seizures were less frequent, febrile in nature, and closely associated with his vaccinations, but were overall controlled by medication. We were diagnosed with Benign Neonatal Familial Epilepsy (BFNE) secondary to KCQN2 genetic mutation in August of 2013. I say we because my entire family had genetic testing and my mother, my brother, my son, and myself were identified with the mutation. My father and John’s father were not.
My mother and I both experienced seizures in infancy, but given the lack of medical information at that time, we were just told that we had seizures. Eventually my mom and I were lucky enough to stop having them after about two years old.
My brother continues to be medicated for seizures, although his last seizure was three years ago. There are no known cognitive deficits for him, my mother, or me that we are aware of.
I knew I had had seizures as in infant and was terrified I would have them during pregnancy, but I hadn’t assumed my son would have a seizure disorder. In retrospect I could have pursued genetic testing prior to becoming pregnant, but I never imagined KCNQ2 existed or had knowledge of the relationship between seizure disorders and genetic mutations. And my doctor was a high-risk pregnancy specialist.
Did you know? In a 2015 study, 27 of 33 families with BFNE had a KCNQ2 mutation. Other implicated genes in the studied population include KCNQ3 and SCN2A.
Searching for supportive services
John is three years old now, and since he turned 18 months old we have been on a journey of heartbreaking developmental issues. It has been a tough month as our private daycare just asked us to leave because it was time to switch groups and they feel John is too developmentally delayed to be integrated with the three-year-olds.
John at one year
At 18 months developmental delays began being investigated by doctors in the neonatology department. John has been identified with a severe speech delay, mild intellectual delay, and has a sensation seeking profile. He is also on a waiting list at the Autism clinic at the Montreal Children’s Hospital.
How are epilepsy and autism related, if at all? Two articles, here and here, describe the complexities and controversies of research in this area.
Privately we have spent our money on speech therapy since last January. Recently we have had a social worker through thecentre local de services communautaires, or local community service centre (CLSC) and an occupational therapist who works with our son weekly. In addition, we are set to begin to receive services at the Mackay Center weekly beginning in January.
My husband and I both have to work full time and we spent more than a month searching for childcare. We were even rejected by a renown special needs pre-school as our son was identified as non-compliant and unworkable. We finally found a private daycare that is costly, and on top of the regular fee they are insisting John does additional therapy at additional cost because of his compliance issues. Unfortunately we cannot be reimbursed by insurance for that as he has no diagnosis of autism (which we do not think he has).
As challenging as his medical issues were, what we are working with now is heartbreaking, but we love our son who is a sweet, affectionate, and kind boy.
Share your KCNQ2 journey here to help raise awareness of KCNQ2-related epilepsy.
Do you ever wonder what it really means to “support research” for KCNQ2? In our first blog, we are happy to introduce you to a special person whose work was one of our earliest investments in KCNQ2 research.
JPF: Please tell us a little bit about what motivates you in your research.
LL: Mutations in the potassium channel gene KCNQ2 cause medically refractory neonatal-onset epilepsy, global developmental delay, and autism. Increasing numbers of clinical cases of KCNQ2 encephalopathy in children have been found in the past couple of years. These cases encourage me to join KCNQ2 study groups.
As a mother to two boys, nothing makes me happier than seeing them smile, watching them laugh, play and grow up healthy. So I understand how hard it is when parents have to face something as hard as watching their child struggle in their development. Being a mother encourages me and gives importance to my goal: To discover good treatments for kids’ diseases using my knowledge and techniques.
.
JPF: Can you please summarize your abstract in lay terms?
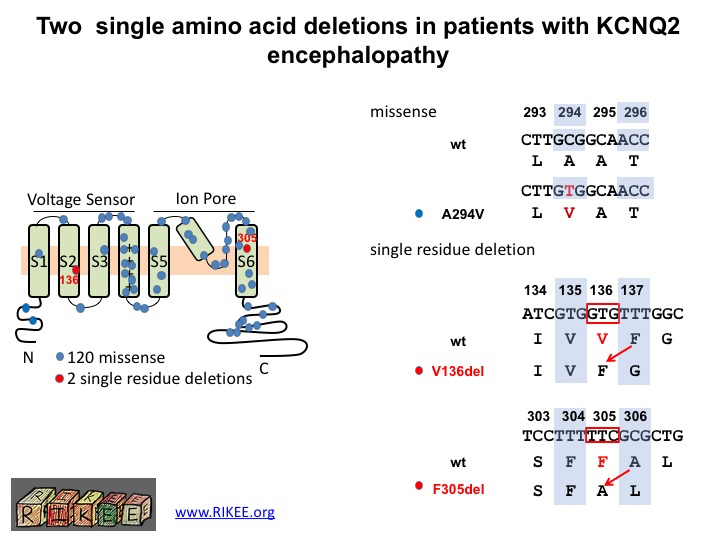
LL: Our lab and collaborators have established an international registry of KCNQ2 cases from patients. To date, we have collected around 120 KCNQ2 encephalopathy missense variants and two interesting examples of single amino acid deletion. One is a unique variant (found in one case), the other in 6 children. My goal was to clarify whether these deletion variants were likely to be the cause of the seizures and impaired development seen in patients.
This diagram shows the location in the KCNQ2 protein of the two newly discovered single amino acid deletions (dots in red) and the 120 previously characterized missense mutations (dots in blue). A missense mutation is a point mutation in which a single nucleotide change results in a codon (a triplet of nucleotides) that codes for a different amino acid. A single amino acid deletion occurs when an entire codon is lost and the rest of the sequence is shifted. Dr. Li’s paper was the first to show that a single amino acid lost, not changed, can induce encephalopathic epilepsy.
JPF: Why do you think your abstract was selected as a highlight? What about your discovery is most valuable in advancing KCNQ2 (or epilepsy) research?
LL: I think my abstract was selected because it is the first time deletion variants have been shown to induce KCNQ2 encephalopathy. I detected strongly reduced channel function and showed a drug could reverse some of the effects of the variant on the channel.
JPF: When did you find out your abstract was selected for special presentation, and how did you prepare for the talk?
LL: In June, I submitted my abstract to the AES annual meeting. After 3 months, in September, I got an email from 2015 the AES scientific program committee. My abstract was selected for a Pediatric Epilepsy Highlights presentation. To prepare, Dr. Cooper and my coworkers gave a lot of valuable advice. It took around a month to organize our clinical and electrophysiological data, and I practiced many times before the presentation.
JPF: Describe your experience of presenting the work. What kind of feedback or questions did you get?
LL: About a thousand people attended my presentation. Most of the audience were doctors. They were very interested in the mechanism and drug treatment. Most of the questions I received were about drug side effects and rules to use drugs to treat encephalopathy, since I presented a new drug (SF0034) that showed similar effects as ezogabine (an FDA approved drug to treat seizures) but which may be less likely to cause the side effects of blue skin discoloration.
JPF: What are you working on next?
LL: I will try to complete these deletion variant studies and publish a paper in the near future. Also I will study other variants in KCNQ2 encephalopathy.
Left to right: Baouyen Tran, a PhD student, Li Li, and Jacquenae Mays, a post-Bac student. Jacquenae and Bao are Li’s collaborators and she thanks them for tirelessly helping her in preparation and rehearsal of her talk.
A native of Dalian, China, Dr. Li Li received her PhD in Department of Pharmacology and Neurobiology at Tokyo Medical and Dental University and her Medical Bachelor degree at Dalian Medical University in Dalian, Liaoning, China. She has studied ion channels for 10 years and joined Dr. Ed Cooper’s Lab at Baylor College of Medicine in 2012 as a postdoctoral research fellow. Her focus is to clarify the mechanisms underlying potassium and calcium channel diseases and develop new drug treatments to cure them
DONATE to The Jack Pribaz Foundation to support research.




 Life with Gillian is unpredictable. As an infant we parented Gillian much in the same way you would any other baby but with the added worry of managing her meds. As she began to grow her inconsistent sleep patterns resulted in severe temper tantrums. This combined with Gillian’s inability to communicate her needs made life difficult at times. Although she is still nonverbal her receptive communication is fairly good and she is better able to understand what we say. Her expressive language is also coming along and we have begun using an iPad to help her communicate.
Life with Gillian is unpredictable. As an infant we parented Gillian much in the same way you would any other baby but with the added worry of managing her meds. As she began to grow her inconsistent sleep patterns resulted in severe temper tantrums. This combined with Gillian’s inability to communicate her needs made life difficult at times. Although she is still nonverbal her receptive communication is fairly good and she is better able to understand what we say. Her expressive language is also coming along and we have begun using an iPad to help her communicate.




 Owen faces down some profound questions when he thinks about his brother’s future. He understands that science takes time, maybe a long time, to come up with the kind of treatment that will help Rhys. He’s “not so happy” about that.
Owen faces down some profound questions when he thinks about his brother’s future. He understands that science takes time, maybe a long time, to come up with the kind of treatment that will help Rhys. He’s “not so happy” about that.