Since last we heard from Molly Turner’s family, there have been big changes for this little girl. And we’re not just talking about her big-surprise-tiny-house! KCNQ2 Journeys is pleased to share this update with readers, who may have first read Molly’s story on the old Jack’s Army website (reprinted below). Our thanks to mom Elizabeth for giving us her insights.

Molly Turner
Q: How old is Molly now?
A: Molly is 6.
Q: How old was your child when diagnosed?
A: Molly was 4 when she was diagnosed.
Q: What would you say have been Molly’s greatest triumphs?
A: Molly continues to amaze our family. She uses a Nova Chat during play to express her wants to us. We couldn’t be more pleased with how she is progressing. She is working on making choices from a set of three objects. Molly has always struggled with her fine motor skills so it’s been a challenging getting her to use her finger to select the correct picture, but we are slowly getting there. Her school has totally embraced the communication device and has offered to send her special ed teacher to training as well. This is a work in progress, but we are so impressed by how much she wants to share with us!
Q: What would you say are your principal challenges with your child? How has that changed since you first shared your story?
A: The biggest challenge with Molly is trying to figure out what she wants and needs since she is nonverbal. When she gets upset it’s very frustrating to both her and me as I don’t know what it is she is upset about. Although this is very difficult for all parties, this is a welcome change. When Molly was younger she was very content with everything. As she’s aged, she’s becoming more aware of her surrounding and craves attention like any child.

Molly the Kosair Kid cover girl
Q: What advice would you give to a parent whose child is newly diagnosed with KCNQ2-related epilepsy?
A: Get support and information early and often! The Facebook page is a great way to keep in touch with other families. I really enjoy bouncing ideas off of other parents.
Q: We can’t help but be a little jealous of Molly’s new digs. Can you tell us the story of her magical playhouse?
A: Molly was fortunate enough to be awarded the Kosair Charities playhouse. Each year Kosair Charities partners with YouthBuild of Louisville and custom builds two playhouses for two special kids in need. There is an application process which includes letter of recommendations collected from therapists and doctors that include how a custom built playhouse would assist in the child’s needs.

A place to play and imagine
See WLKY news coverage of Molly’s playhouse arriving at her house.
Molly’s playhouse is a special place she can go to spend time with her siblings and friends where she can just be a kid. As Molly has aged, Chris and I find it difficult to integrate Molly’s likes into activities that she and her siblings can do together since her needs are so great and her lack of mobility makes it hard for her to be active. Molly’s playhouse has a TV, DVD player, puzzles, dress up clothes, a book nook, bubbles, mirrors, anything a 6-year-old would want–all in our backyard.
What’s your child’s KCNQ2 story? Share it here to help raise awareness of KCNQ2-related epilepsy.
Molly’s original story
On July 30, 2009, Molly was born at Baptist East Hospital in Louisville, Kentucky. Molly was breech, so she was delivered via a very uneventful, standard Caesarean birth. Initially, Molly seemed a perfect 7lb, 21 inch bouncing baby girl, and we were overjoyed with our now family of five.
During her first night, Molly became very sleepy and lost interest in breastfeeding. On the 31st, she started shaking, but the nurses thought it was just low blood sugar because she was not eating well. Around Noon that day, Molly had her first documented seizure and was immediately transferred downtown to Kosair Children’s Hospital. Her labs, MRI and CAT scan all came back normal, but the seizures didn’t stop.
A regimen of Phenobarbital, Keppra and Dilantin was started, but the seizures kept coming.
Molly spent nearly a month highly sedated from the medication and on a feeding tube in the Kosair NICU while the doctors tried everything they could to get the seizures under control. Some days were better than others, but the wonderful, caring people we met during our stay at Kosair helped us every step of the way and has become a phenomenal network.
Molly finally got to come home September 3, 2009, with a diagnosis of KCNQ2 mutation, with the hope that her seizures would stop and she would develop normally. The first six months of Molly’s life were spent in and out of Kosair trying to control the seizures. Molly’s EEGs continued to show seizure activity, so her neurologist weaned her off Dilantin and Keppra and started the medication Trileptal.
When Molly began having delays in her development, her neurologist and geneticist started looking into other genetic mutations because most children in 2009 and before that were diagnosed with the KCNQ2 mutation did not exhibit the delays she was presenting.
We started seeing other specialists outside of Kentucky and visited The Cleveland Clinic where the neurologist we met with discounted the KCNQ2 mutation as well and tested Molly for other mutations and syndromes. This continued until the fall of 2013 when we decided to receive gene-sequencing test on Molly, my husband Chris and myself. The results came back in May 2014, with de novo KCNQ2 mutation only. After additional research, our neurologist confirmed that there are more severe cases that present with significant developmental delays. That very afternoon we found a new support group in Jack’s Army!
Molly continues to get therapy both at Harmony Elementary and The Kids Center in Louisville, Kentucky. She continues to struggle with feeding, but can eat a mashed/puréed diet. She is not verbal or mobile. She has cortical visual impairment and has had surgery for her strabismus.
Molly is a beautiful addition to our family. Without her presence, I wouldn’t have met some of the most wonderful nurses, doctors, therapists, special education teachers and special needs parents in the world. Along with our family and friends, these individuals are truly are lifeline and support.
Molly allows me to see the ability in her disabilities every day, and I thank God for that gift.





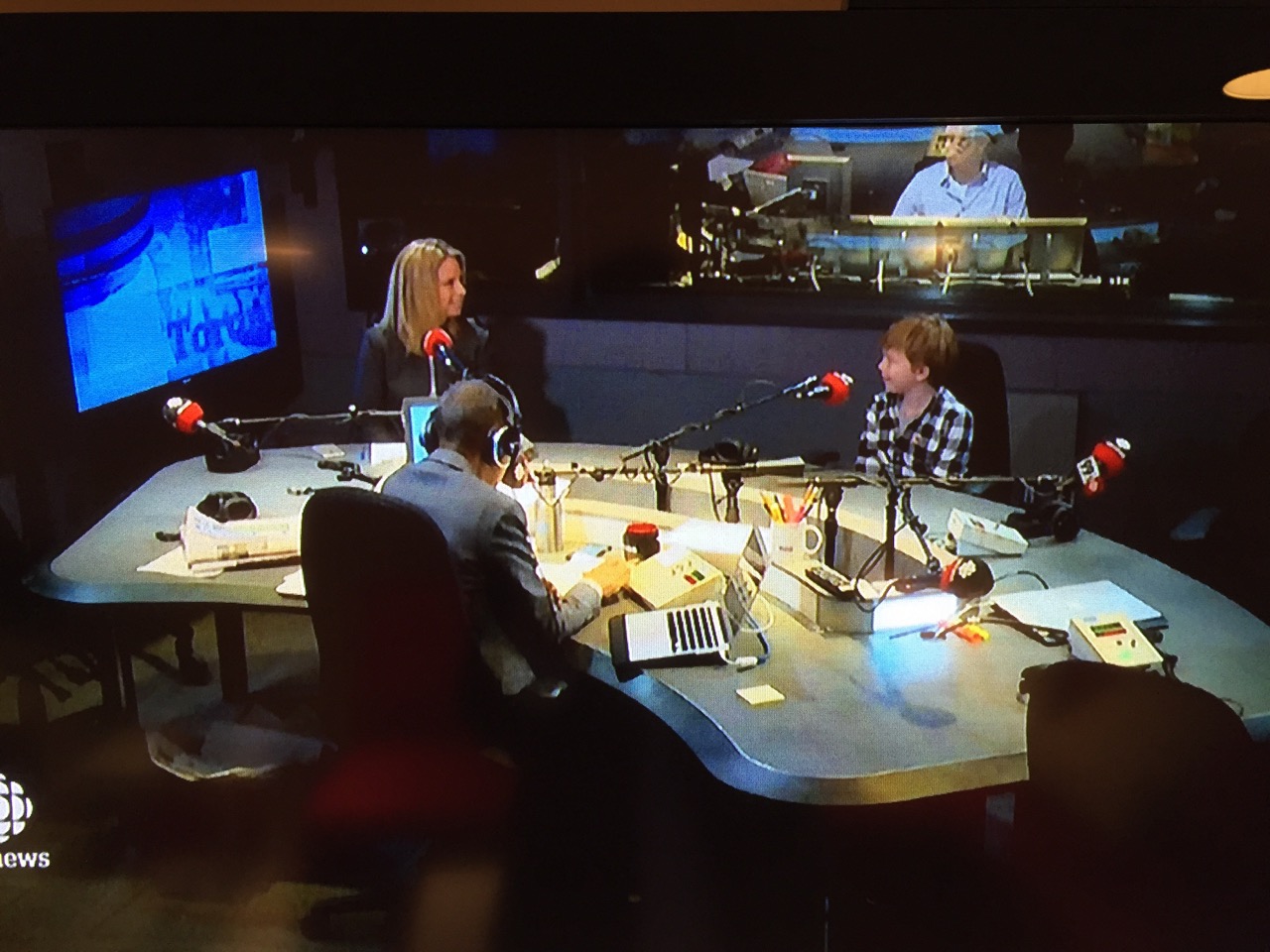

 Owen faces down some profound questions when he thinks about his brother’s future. He understands that science takes time, maybe a long time, to come up with the kind of treatment that will help Rhys. He’s “not so happy” about that.
Owen faces down some profound questions when he thinks about his brother’s future. He understands that science takes time, maybe a long time, to come up with the kind of treatment that will help Rhys. He’s “not so happy” about that.